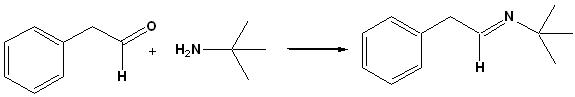NMR Overlay with JSpecView
Click here to start an applet with an example of spectral overlay using JSpecView.
Robert Lancashire updated the code for JSpecView to fix the problem of faulty peak offset for NMR spectra in overlay mode. I have updated our server so that the new applets will be downloaded for UsefulChem applications. The new JAR files are available from Sourceforge.
It is hard to explain in words just how powerful this is for monitoring reactions and providing useful minable data in an Open Chemistry environment. So I made a recording to show how it can be used to monitor the formation of the imine in EXP045, the first step of the Ugi reaction. By using enough superimposed spectra from key points in the reaction, it is possible to resolve an ambiguous interpretation of an NMR showing only the final result.
The merging of spectra is accomplished easily by creating a BLOCK file from individual JCAMP files:
1) Create at txt file using Notepad and rename it filename.jdx
2) Add this header
##TITLE=IMINE FORMATION
##JCAMP-DX= 5.01
##DATA TYPE= LINK
3) Add this footer
##END= $$end of BLOCKs
4) In between the header and footer insert single JDX files by opening them in Notepad then copying and pasting. It is ok to leave space between the file blocks for clarity.
5) The first parameter of each file block is ##TITLE=. Put something meaningful there because this will be used by the overlay key to identify each spectrum by color.
6) Download this html file by right clicking and saving. Modify the 3 places in the file that call the JDX file to link to the one you just created.
7) Upload the JDX and HTML files to a server with the new JSpecView JAR files in the same folder.
8) Link to the HTML file to use JSpecView in overlay mode. Firefox tends to crash when going back and forth between applets and needs to be reloaded to fix that.
It is important that all spectra be zeroed on TMS properly. If a spectrum is off, use JSpecView to calculate how far off in Hertz (ppm * MHz of the NMR, in our case 300). To shift the spectrum to the left (downfield) subtract this correction factor from the number under the parameter REFERENCE_POINT= in the JDX file.
1/C6H9NO/c1-5-2-3-6(4-7)8-5/h2-3H,4,7H2,1H3
5-methylfurfurylamine
posted by Jean-Claude Bradley @ Friday, December 29, 2006
0 comments
![]()