



Automated Reaction Kinetics using Excel VBA and JCAMP
Dave has created the required files and code to substantially automate the integration processing of NMR files in order to extract kinetic information. Two weeks ago I suggested that we use CML to do this but for now it is a bit more convenient to use JCAMP BLOCK files.
Here is the procedure:
1) Uncompress the JCAMP jdx files obtained from the NMR by loading them one by one in the standalone version of JSpecView and exporting as JCAMP XY format. In the TITLE field (first line) replace the default name with the number of minutes after the addition of the last reagent or the name of the starting material.
2) Create a new txt file and affix a .jdx extension. At the start of the file insert the following lines:
##TITLE= Reaction Profile Title
##JCAMP-DX= 5.01
##DATA TYPE= LINK
and at the end of the file add:
##END= $$end of BLOCKs
3) In between these lines insert the uncompressed JCAMP files created above. Empty lines to separate the spectra are ok.
4) Download and extract the following zip folder and modify the JCAMPNMR.ini file according to the number and location of ranges. The integration ranges should be selected carefully to ensure that the peaks will not drift out of range while being narrow enough to minimize baseline drift issues. (Leave the Frequency at 500 for now even if using the 300 MHz instrument - it works like it is.)
5) Open the JCAMPNMR.xls Excel workbook in the same folder as the ini file. You should have your Excel security settings set to allow macros. If it is working ok Excel will ask you to choose a file. Browse to the BLOCK file created above. Give it a few minutes to process and it should produce a table of all integrations for the ranges set in the ini file for all spectra in the BLOCK file.
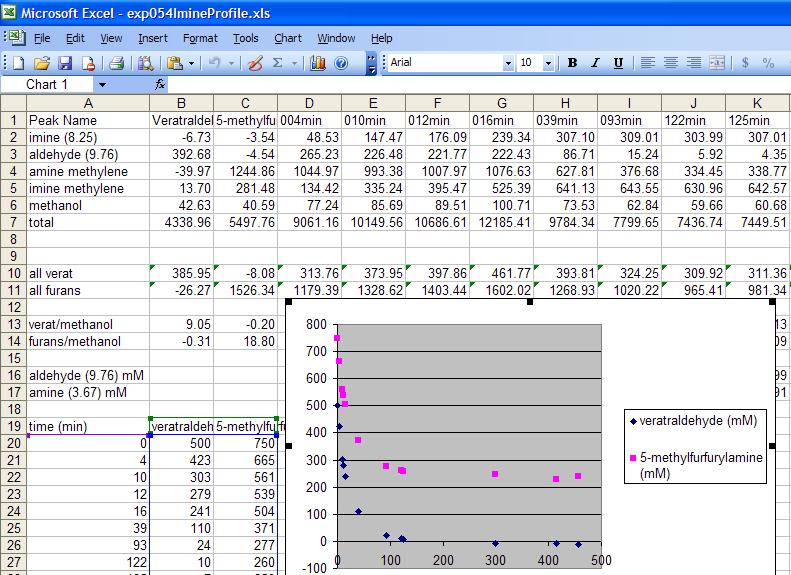
The exp054ImineProfile.xls file included in the zip folder is an example of a processed BLOCK file with 11 monitoring spectra and 2 starting materials for the formation of an imine in EXP054. The plot in the file shows the decrease in the concentration of the aldehyde and amine as the imine is formed. It appears that 50% excess amine was added in this experiment thus the aldehyde concentration goes to zero while the amine ends at 250 mM. The assumption used in this plot is that the only significant process occurring here is the formation of the imine.
So there is still some thinking and common chemical sense required but the researcher does not have to manually perform the integrations of several peaks on 13 spectra, which I can attest is tedious and prone to error. I am hoping that providing these data openly in such a format will encourage collaboration and further automated processing. For example I am a bit rusty on fitting second order kinetics to reactions with different starting concentrations of reactants. Anybody want to help with that?
The resulting workbook also contains each spectrum with integration as a separate worksheets. There appears to be more drift in the baseline than observed with JSpecView so I'll have to contact Robert Lancashire to see if there is some trick to that. We are just doing a simple summation of data points. That is why there are small negative numbers in the integration dataset.
In principle the BLOCK files should be viewable with JSpecView as overlays. However, with 13 spectra, that maxes out the Java memory, even with the enhancement to 200 Meg. If some spectra are deleted the overlay should work at some point, depending on the computer's Java memory.
The most time consuming task now is uncompressing the JCAMP files. If we learn how to do that automatically and if the code could read the time off of the filenames and write in the titles, then creating the BLOCK file could be as simple as dragging a bunch of jdx files in a folder and clicking a button to activate an Excel macro.
At some point if we (or anyone) can add intelligent peak picking, we may be able to have some bots analyze the reaction course of ANY reaction profile as a routine matter and flag kinetic behaviors of potential interest.
posted by Jean-Claude Bradley @ Sunday, January 28, 2007
2 comments
![]()

2 Comments:
Here's a cool spreadsheet that simulations reaction kinetics in Excel by solving the discretized differential equations
thanks Jane - looks like a useful package
Post a Comment
<< Home